Narzędzia chirurgiczne i narzędzia stomatologiczne w świetle obecnych oraz planowanych regulacji prawnych

Narzędzia chirurgiczne i narzędzia stomatologiczne– obecne przepisy
Wytyczne unijne dla wyrobów medycznych reguluje Dyrektywa Rady 93/42/EWG z dnia 14 czerwca 1993 r. W Polsce dyrektywa ta jest zaimplementowana przez ustawę z dnia 20 maja 2010 r. o wyrobach medycznych (Dz.U. z 2015 r., poz. 876 ze zmianami).
Uzupełnieniem ustawy o wyrobach medycznych są rozporządzenia Ministra Zdrowia, określające szczegółowe wytyczne w zakresie: klasyfikacji wyrobów medycznych, zasad przeprowadzania oceny zgodności, sposobów powiadamiania i zgłaszania o wprowadzeniu do obrotu, zasad ubiegania się o zgodę na przeprowadzenie badań klinicznych.
Jednostką sprawującą nadzór nad przestrzeganiem Ustawy o wyrobach medycznych jest w Polsce rzędu Rejestracji Produktów Leczniczych, Wyrobów Medycznych i Produktów Biobójczych. Dodatkowo wyroby medyczne mogą w zależności od ich klasyfikacji podlegać nadzorowi jednostki notyfikowanej, która po spełnieniu przez producenta określonych wymagań, wystawia certyfikat CE.
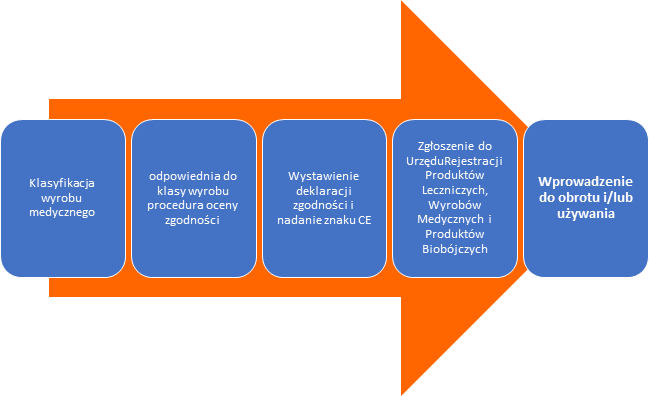
Ścieżkę wprowadzania wyrobu do obrotu lub/i używania na terenie Polski przedstawia następujący schemat:
Klasyfikacja wyrobów medycznych
Klasyfikacja wyrobu medycznego jest bardzo ważna. Od niej zależy rodzaj procedury oceny zgodności, którą ma wykonać wytwórca, aby zapewnić, że oceniany wyrób spełnia wymagania zasadnicze. Im wyższa klasa wyrobu, tym bardziej restrykcyjna procedura oceny zgodności.
Wyróżniamy 6 klas wyrobów medycznych
klasa I (np. kołnierze ortopedyczne, rękawice do badań, wózki inwalidzkie, narzędzia chirurgiczne wielokrotnego użytku),
klasa I – wyroby z funkcją pomiarową,
klasa I – wyroby sterylne,
klasa IIa (np. opatrunki hydrożelowe, cewniki jednorazowe, klisze rentgenowskie),
klasa IIb (np. pojemniki na krew, prezerwatywy, respiratory),
klasa III (np. implanty piersi, zastawki serca, protezy naczyniowe).
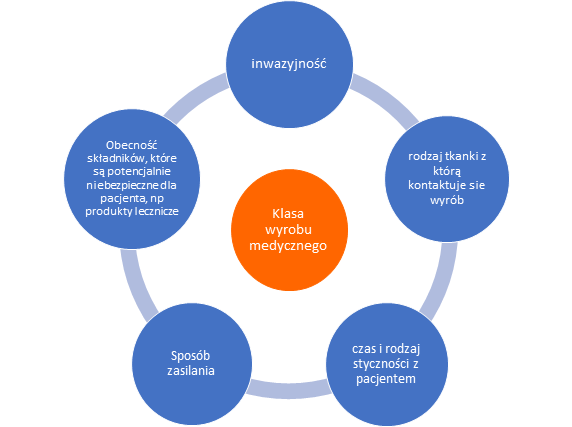
W klasyfikacji wyrobów medycznych bierze się pod uwagę szereg czynników takich jak:
Należy też pamiętać, że narzędzie chirurgiczne wielokrotnego użytku ma w Ustawie o wyrobach medycznych swoją definicję:
„Narzędzie chirurgiczne wielokrotnego użytku” oznacza narzędzie przeznaczone do użytku chirurgicznego: cięcia, wiercenia, piłowania, drapania, skrobania, zaciskania, odciągania, klamrowania lub do podobnych zabiegów, bez podłączenia do jakiegokolwiek aktywnego wyrobu, które to narzędzie przewidziane jest przez producenta do ponownego użytku po przeprowadzeniu odpowiednich procedur, takich jak czyszczenie, dezynfekcja i sterylizacja.
Czyli np. narzędziem chirurgicznym będą z pewnością: pincety, nożyczki chirurgiczne, skalpele, hak chirurgiczny, ale np. ostrze piły zasilanej elektrycznie lub wiertło do wiertarki elektrycznej nie będzie się już mieściło w tej kategorii (będzie sklasyfikowane wyżej).
W świetle obecnych przepisów (wg Dyrektywy Rady 93/42/EWG) procedura oceny zgodności dla wyrobów medycznych klasy I (za wyjątkiem wyrobów sterylnych i z funkcją pomiarową) nie wymaga udziału jednostki notyfikowanej. Producent po przeprowadzeniu oceny zgodności i sporządzeniu dokumentacji technicznej wystawia deklarację zgodności i oznakowuje wyrób znakiem CE oraz rejestruje wyrób w Urzędzie Rejestracji.
Wyroby pozostałych klas inne niż wyroby wykonane na zamówienie, wyroby do badań klinicznych, wyroby do oceny działania i wyroby wykonane przez użytkownika wymagają udziału Jednostki Notyfikowanej do przeprowadzenie procedury oceny zgodności w zakresie zależnym od klasy wyrobu. W większości przypadków ( za wyjątkiem tzw, weryfikacji WE) wymagane jest również wdrożenie Systemu Zarządzania Jakością (ISO 13485).
Po wystawieniu certyfikatu CE przez jednostkę notyfikowaną, producent wystawia deklaracje zgodności oraz oznakowuje wyroby znakiem CE z numerem jednostki notyfikowanej.
Procedura oceny zgodności wyrobu medycznego obejmuje również sporządzenie dokumentacji technicznej wyrobu, która zostanie omówiona szczegółowo w następnym artykule.
Planowane zmiany dotyczące klasyfikacji i procedur oceny zgodności dla narzędzi chirurgicznych- MDR 2017/745
Regulacje prawne dla wyrobów medycznych na rynku europejskim zmienia Rozporządzenie unijne (UE) 2017/745 z 5 kwietnia 2017 r. (Medical Device Regulation – MDR). Wymagania będą stopniowo wprowadzane aż do 2025 roku. Większość wymagań będzie miało zastosowanie od dnia 26.05.2021 (termin przesunięty o 1 rok z powodu pandemii koronawirusa).
Jeżeli chodzi o narzędzia chirurgiczne wielokrotnego użytku klasy I (określane symbolem Ir), zgodnie z MDR procedura oceny zgodności przebiega z udziałem jednostki notyfikowanej, co w zależności od wybranej ścieżki oceny zgodności, wymaga od producenta posiadania Systemu Zarządzania Jakością spełniającego określone wymagania. Jest to w porównaniu z obecnymi przepisami dość spora rewolucja dla tego typu wyrobów (według MDD 93/42/EEC udział jednostki Notyfikowanej nie jest wymagany).
W wyniku wprowadzenia poprawki do MDR, producenci narzędzi chirurgicznych wielokrotnego użytku, zyskali dodatkowy czas na przygotowanie się do nowych wymagań tj. do 26 maja 2024. Warto jednak już teraz przygotować się do nadchodzących zmian.
Narzędzia wprowadzane do obrotu po tym czasie muszą posiadać znak CE z numerem jednostki notyfikowanej.
Nadzór jednostki notyfikowanej na narzędziami chirurgicznymi wielokrotnego użytku klasy Ir sprowadza się głównie do aspektów wielokrotnego użycia, czyli czyszczenie, dezynfekcja, sterylizacja, konserwacja, testowanie funkcjonalne oraz odpowiednie instrukcje używania.
UWAGA: ograniczony zakres nadzoru jednostki Notyfikowanej nie zwalnia producenta z obowiązku sporządzenia pełnej dokumentacji technicznej wyrobu.
Dodatkowo Jednostka Notyfikowana ocenia System Zarządzania Jakością wdrożony przez producenta. W zależności od wybranej drogi oceny zgodności producent może wdrożyć Pełny System Zarządzania Jakością lub System Zarządzania Jakością zatwierdzony do produkcji danych wyrobów.
Producent narzędzi chirurgicznych musi się liczyć również z możliwością niezapowiedzianego audytu, który jednostka notyfikowana przeprowadza przynajmniej raz na 5 lat.